
The survey results are in…Medical Devices - Quality Management System
Key findings from NSAI national user survey on the need for changes
ISO 13485:2016 – Medical Devices – Quality Management Systems – Requirements for regulatory purposes is an important standard for the medical device industry.
The standard outlines the requirements for a Quality Management System (QMS) that enables organisations to consistently provide medical devices that meet customer and applicable regulatory requirements.
Since its last revision, the standard has gained global recognition by regulatory authorities in Europe, USA, Canada, Australia and Japan. Certification to the current version signifies a commitment to quality and regulatory compliance.
Systematic review
ISO/Technical Committee 210, responsible for quality management and related aspects for health products including medical devices, developed the standard. As part of the ISO Standard development process, all published standards undergo a review every 5 years.
The systematic review of ISO 13485 recently took place with feedback on possible changes due by June 6th, 2025. All ISO member bodies were invited to gather user feedback on potential changes.
Feedback from Ireland
As part of this process, members of NSAI/TC 5 Healthcare Standards Consultative Committee developed a user survey to gather national feedback on any possible changes that may be required. The NSAI survey ran for approximately 6 weeks from early March until mid-April, 2025.
The survey was available on the NSAI website and was highlighted to members of the Irish Medtech Association, companies certified to the standard through NSAI and national experts participating in standards development committees. A total of 51 responses were received and the results are summarised below.
Survey results
Systematic review of ISO 13485 – feedback and survey responses
Should the standard be revised or kept the same?
A key question for the ISO systematic review process is asking if the standard needs to be revised or kept the same.
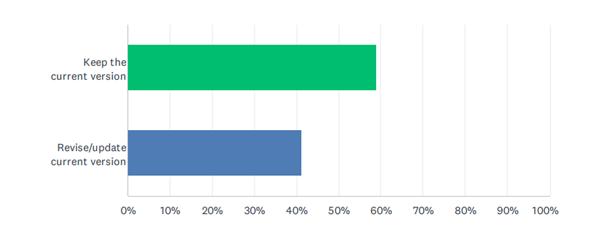
NSAI’s survey results revealed that 59% of respondents preferred to keep the standard the same, while 41% supported revision of the current version – see figure 1.
Figure 1: Survey responses to Systematic review of ISO 13485
Many of the respondents provided a reason for their answer.
The global recognition that the standard has received from different regulatory jurisdictions (FDA, EU, Canada, MDSAP) was a key reason for implementing no change at this time.
Many stated that current version provides sufficient details for a Quality Management System with a focus on safe manufacturing of medical devices. Any changes implemented now would not bring any additional benefits to manufacturers who are currently working towards compliance to new regulations such as EU Medical Device Regulation.
While some indicated that changes were needed, most were seeking clarity and further guidance, particularly for contract manufacturers who may not be responsible for placing products on the market.
With the development of complex supply chains, supplier controls and responsibilities were areas where more guidance would be welcomed. Better alignment with the European regulations and requirements for quality management systems was also suggested.
The current standard is still quite focused on traditional production and product manufacturing and therefore, guidance on how the standard applies to medical device software and servicing activities was also suggested. The feedback loop from post-market surveillance and post-market clinical feedback into product change and development could be better addressed in the standard, according to the survey results.
Clauses highlighted that may require change:
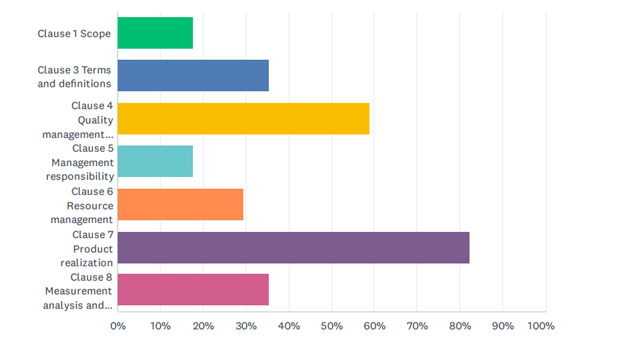
When asked which clauses of the current standard should be revised, all clauses were indicated, although two specific clauses clearly ranked the highest – see Figure 2.
Survey responses indicated that nearly 60% of respondents believed that Clause 4 (Quality management system) required updating while 82% favoured clause 7 (Product realization).
Figure 2: Clauses of ISO 13485 where change maybe required
Feedback on particular changes in these clauses indicated that Clause 4 should address the need for a medical device file/technical file and maintenance of such data.
Control of records in 4.2.5 should better reflect digitisation and should provide guidance on retention times for all records, including digital records, according to several responses.
Better clarity on how the standard addresses risk for both product (ISO 14971) and process, (what QM is about) was also highlighted.
The flow of Clause 7 was reported as not being user friendly with too much focus on traditional product manufacturing. The application to more service or software development organisations may not always follow the current standard and was an area suggested for improvement.
How to adequately address both product and process risk was also an area some reported as requiring improvement.
Alignment with other management system standards, such as ISO 9001, was also suggested, as many organisations may have to apply both standards. This conflicted with other respondents however, who specifically indicated that the standard should not be aligned with ISO 9001.
A reason for this difference in opinion could be that ISO 13485 has a very specific regulatory application and moving to the common management system approach may go beyond what is necessary for regulatory purposes.
The survey also included a question about developing a guidance document on the application of ISO 13485:2016 to assist the medical device industry.
92% of respondents were in favour of the development of such a document, with only 7% indicating no need for such a document.
The development of a guidance document could assist by providing examples and case studies to show how particular issues can be interpreted and applied for different users (e.g. contract manufacturers).
Many of the same topics were reported here for guidance, as was suggested for possible changes to the standard, including:
-
risk management for product and processes
-
application to software as a medical device and app developers
-
outsourcing and relevant responsibilities and controls
-
post-market surveillance and the feedback loop into product development could be better addressed
-
guidance on corrective actions and preventive actions and how to address these was suggested – particularly as preventive actions are no longer used in other management system standards.
Respondent demographics
Type of organisation:
Nearly half of respondents (47%) represented medical device manufacturers followed by consultants (27.4%) accreditation bodies (7.8%), regulators (7.8%) and distributors of medical devices (5.8%). The remainder included auditor, medical device packaging and a notified body. Medical manufacturers, as the primary users of the standard constituted the largest stakeholder group.
Size of Organisation:
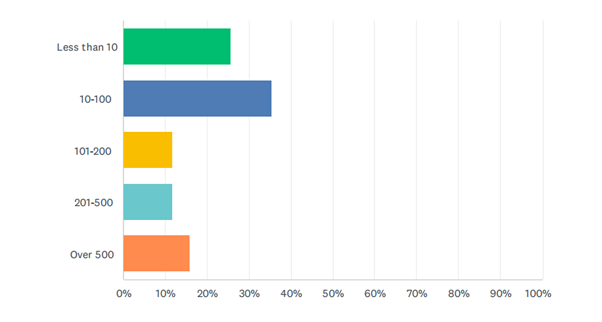
Almost 61% worked in an SME (up to 100 employees), while 24.5% worked in organisations with 101-500 employees and 15.7% worked in larger organisations (over 500).
See figure 3 for more information:
Figure 3: Survey respondents - Size of organisation
Roles of respondents
The majority of respondents were Quality/Regulatory managers (33.3%) and consultants (25.5%), followed by auditors (15.7%) and regulatory assessors (7.8%). Other roles included manufacturing manager, suppliers, and various engineering roles such as R&D, Quality, risk and design assurance.
Relevant Market areas
The survey asked respondents to indicate all market areas that were relevant to their organisation.
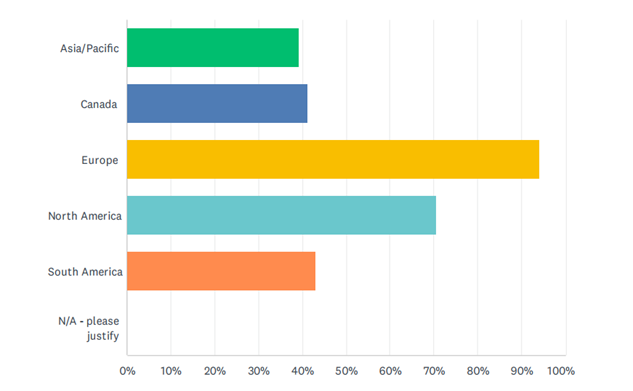
Europe was the main market area of interest (94%) followed by North America (71%). South America, Canada were almost equal (43% & 41% respectively). Asia/Pacific had the lowest level of interest among the respondents (39%). See Figure 2 for more information.
Figure 4: Market areas relevant to survey respondents
Conclusions
The results of the survey indicate that most users of ISO 13485 are content with the current standard and support its confirmation during the systematic review.
However, some sections of ISO 13485 particularly Clause 7 (Product realization) and Clause 4 (Quality Management) were identified as problematic. This may be due to the shift to development of medical device software and healthcare apps, as well as the use of complex contract manufacturing and supply chains.
Most respondents strongly favoured the development of a guidance document on the application of ISO 13485. ISO is currently working on a new Technical Specification – ISO TS 23458 – Medical devices – Quality management systems – Guidance for the application of ISO 13485:2016.
Next steps
NSAI will used the results from this survey as part of its feedback to ISO on the systematic review of ISO 13485, which should inform the development of the new guidance document.
If you’d like to find out more about the work of NSAI/TC 5 Healthcare Standards Consultative Committee, please email info@nsai.ie with ‘NSAI/TC5’ in the subject line.
To view or download standards associated with medical devices, please visit NSAI’s webstore at www.standards.ie
[Disclaimer: All reasonable effort was made to ensure that the information on this page was correct at the time of publication. Any views or opinions expressed on this page are not necessarily those of NSAI. NSAI accepts no responsibility or liability howsoever arising from the contents of this publication or any errors, inaccuracies, or omissions in the contents of the information provided therein.]


