
NSAI Publishes Revised Risk Management Standard for Medical Devices - I.S. EN ISO 14971:2019
A new revision of the medical device risk management standard – I.S. EN ISO 14971:2019 has been published by NSAI and is available from NSAI standard publications – www.standards.ie.
I.S. EN ISO 14971:2019 is the adopted Irish Version of the European Document EN ISO 14971:2019, Medical devices, Application of risk management to medical devices (ISO 14971:2019).
This document supersedes EN ISO 14971:2012 and ISO 14971:2007
The new edition does not contain Annex Z’s demonstrating the relationship with relevant European Regulations and therefore is not Harmonized i.e. giving presumption of conformity to EU regulations. The standard will be updated with an amendment to add the content of the Annex Z’s once they are finalised by CEN/CLC/JTC3.
Reason for Change
The changes aim to deal with lessons learned and take account of new and emerging risks from a wide range of medical devices in the market including software, healthcare applications, electrical equipment, etc. The standard now acknowledges risks outlined in other standards depending on the device type (ISO 60601-1, ISO 62366, ISO 62304, ISO 27000, etc).
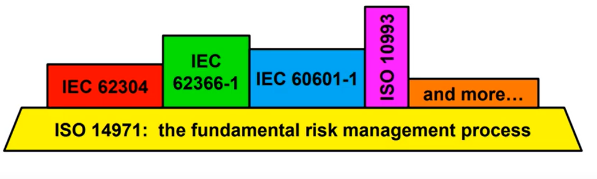
Figure 1 Representation of ISO 14971 with additional standards for dealing with risk
To that end, EN ISO 14971:2019 is deemed to be the fundamental framework for product risk management working together with alternative standards which may call out specific risks.
Hence, there are many significant changes made under this revision to support the development of safe and effective medical devices, starting with the introduction of a new clause (refer to Table 1).
Table 1 Structural Differences Between 14971 current and previous standards
|
Clause |
ISO 14971:2007 EN ISO 14971:2012 |
Clause |
ISO 14971:2019 (3rd Edition) |
|
1 |
Scope |
1 |
Scope |
|
2 |
Terms and Definitions |
2 |
Normative Reference |
|
3 |
General Requirement for Risk Management |
3 |
Terms and Definitions |
|
4 |
Risk Analysis |
4 |
General Requirement for Risk Management System |
|
5 |
Risk Evaluation |
5 |
Risk Analysis |
|
6 |
Risk Control |
6 |
Risk Evaluation |
|
7 |
Evaluation of Overall Residual Risk Acceptability |
7 |
Risk Control |
|
8 |
Risk management Report |
8 |
Evaluation of Overall Residual Risk |
|
9 |
Product and Post Product Information |
9 |
Risk Management Review |
|
|
|
10 |
Production and Post Production Activities |
|
|
|
|
Annex A: Rationale for Requirements |
|
|
|
|
Annex B: Risk Management process for medical devices |
|
|
|
|
Annex C: Fundamental risk concepts |
The main changes compared to the previous edition are as follows:
- A clause on normative references has been included, in order to respect the requirements for fixed in Clause 15 of ISO/IEC Directives, Part 2:2018.
- The defined terms are updated, and many are derived from ISO/IEC Guide 63:2019. Defined terms are printed in italic to assist the reader in identifying them in the body of the document.
- Definitions of benefit, reasonably foreseeable misuse and state of the art have been introduced.
- More attention is given to the benefits that are expected from the use of the medical device. The term benefit-risk analysis has been aligned with terminology used in some regulations.
- It is explained that the process described in ISO 14971 can be used for managing risks associated with medical devices, including those related to data and systems security.
- The method for the evaluation of the overall residual risk and the criteria for its acceptability are required to be defined in the risk management plan. The method can include gathering and reviewing data and literature for the medical device and for similar medical devices and similar other products on the market. The criteria for the acceptability of the overall residual risk can be different from the criteria for acceptability of individual risks.
- The requirements to disclose residual risks have been moved and merged into one requirement, after the overall residual risk has been evaluated and judged acceptable.
- The review before commercial distribution of the medical device concerns the execution of the risk management plan. The results of the review are documented as the risk management report.
- The requirements for production and post-production activities have been clarified and restructured. More detail is given on the information to be collected and the actions to be taken when the collected information has been reviewed and determined to be relevant to safety.
- Several informative annexes are moved to the guidance in ISO/TR 24971, which has been revised in parallel and expected to be released in 2020. More information and a rationale for the requirements in this third edition of ISO 14971 have been provided in Annex A. The correspondence between the clauses of the second edition and those of this third edition is given in Annex B.
For queries or questions, contact info@nsai.ie.
[Disclaimer: All reasonable effort was made to ensure that the information on this page was correct at the time of publication. Any views or opinions expressed on this page are not necessarily those of NSAI. NSAI accepts no responsibility or liability howsoever arising from the contents of this publication or any errors, inaccuracies, or omissions in the contents of the information provided therein.]


